
「禁忌を含む注意事項等情報」等については、製品情報(DI)をご参照ください。
国内第III相試験
(FSN-013P-03試験)
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
試験概要
- 目 的:
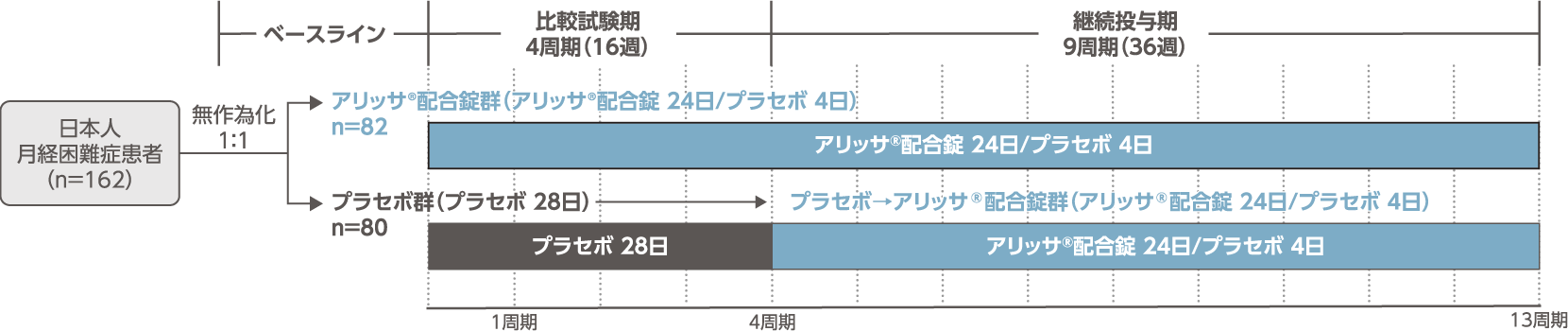
- 日本人月経困難症患者を対象に、アリッサ®配合錠を4周期(16週間)投与した際の月経困難症に対する有効性について、プラセボに対する優越性を検証する。また、13周期(52週間)投与した際の長期安全性を検討する。
- デザイン:
- 多施設共同、無作為化、二重盲検、プラセボ対照、並行群間比較試験
- 対 象:
- 日本人月経困難症患者162例
- 方 法:
- 治験薬を1日1回1錠、毎日原則一定の時刻に経口投与する。患者は以下の各群に1:1で無作為に割付けられた。
- [アリッサ®配合錠群]
- アリッサ®配合錠を24日間投与した後にプラセボを4日間投与する。計28日間を1周期として、4周期投与(比較試験期)と9周期投与(継続投与期)の計13周期とした。
- [プラセボ群]
- プラセボの28日間投与を1周期とし、4周期投与する(比較試験期)。その後、アリッサ®配合錠を24日間投与した後にプラセボを4日間投与する計28日間を1周期とし、9周期投与する(継続投与期)。投与期間は計13周期とした。

- 評価項目:
-
- [主要評価項目]
(検証的な解析項目) - 月経困難症スコアのベースラインから4周期目までの変化量
なお、ベースラインは、無作為化の直前2回の月経周期における月経困難症スコア合計値の高い方とする。
- [副次評価項目]
- 月経困難症スコア・月経困難症症状に対するVAS(visual analogue scale)値・月経時又は消退出血時の骨盤痛(下腹部痛・腰痛)VAS値・月経時以外又は消退出血時以外の骨盤痛スコア・月経時以外又は消退出血時以外の骨盤痛(下腹部痛・腰痛)VAS値のベースラインからの変化量、血清ホルモン濃度(血清エストラジオール濃度、血清プロゲステロン濃度、血清FSH濃度、血清LH濃度)、ダグラス窩の硬結、子宮可動性の制限、骨盤の圧痛、日常活動に対する障害、睡眠に対する障害、臨床的全般改善度、臨床的全般満足度 など
- [安全性評価項目]
- 有害事象 など
- [主要評価項目]
- 解析計画:
-
- [主要評価項目]
- 最大の解析対象集団(無作為化され、治験薬を少なくとも1回以上投与された患者、FAS)を対象とした。アリッサ®配合錠群及びプラセボ群の投与群別に平均値±標準偏差の推移図を作成し、平均値の両側95%信頼区間を算出した。また、月経困難症スコアのベースラインからの変化量は、共分散分析(ANCOVA)※を用いて、両群間の差の最小二乗平均値及び標準誤差を算出した。治験薬投与開始後の当該評価時点に評価可能なデータが存在しない場合(欠測値)にはLast observation carried forward(LOCF)法を適用し、それ以前の評価可能なデータのうち、最も近いものを当該評価時点のデータとして採用した。
※ANCOVA:月経困難症スコアの変化量を従属変数とし、投与群及び月経困難症診断カテゴリー(機能性、器質性)を固定効果、ベースラインの月経困難症スコアを共変量とした(有意水準:片側2.5%)。
- [副次評価項目]
- FASを対象とした。アリッサ®配合錠群及びプラセボ群の連続変数については、計量値及びベースラインからの変化量の要約統計量を投与群別に評価時点毎に算出した。また、分類変数については、頻度集計を投与群毎に行った。
月経困難症症状に対するVAS値、月経時又は消退出血時の骨盤痛(下腹部痛・腰痛)VAS値、月経時以外又は消退出血時以外の骨盤痛スコア及び月経時以外又は消退出血時以外の骨盤痛(下腹部痛・腰痛)VAS値は、ベースラインから4周期目までの変化量を主要評価項目と同様に解析した。
欠測値には、主要評価項目と同様にLOCF法を適用した。
- [安全性評価項目]
- 安全性解析対象集団(治験薬を少なくとも1回以上投与された患者、SAS)を対象に、MedDRA ver.24.0を使用した。特に注目すべき有害事象として、血栓症(血栓塞栓症を含む)に関連する有害事象、エストロゲン・プロゲスチン(EP)配合剤投与に関連すると考えられるその他の有害事象、ドロスピレノン(DRSP)投与に関連すると考えられる有害事象、器質性疾患に関連する有害事象を設定した。
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
組み入れ基準
- 選択基準:
-
- (1)器質性月経困難症又は機能性月経困難症と診断された日本人患者
<器質性月経困難症>
開腹又は腹腔鏡検査により子宮内膜症又は子宮腺筋症と確定診断された患者
経腟超音波検査(transvaginal ultrasonography: TVUS)又は核磁気共鳴画像法(MRI)により子宮内膜症(卵巣子宮内膜症性囊胞を認める)、子宮腺筋症又は子宮筋腫と診断された患者
<機能性月経困難症>
問診、内診及びTVUSなどにより、器質性疾患が否定された患者 - (2)同意取得時の年齢が20歳以上の患者
- (3)無作為化割付け前、直前2回の月経時において月経困難症スコアの合計がいずれも3点以上の患者
- (4)無作為化割付け前、直前2回の月経周期が25~38日の患者
- (5)Body mass index(BMI)が30kg/m2未満の患者
- (6)本治験の内容を十分理解し、治験への参加を文書にて同意した患者
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
- (1)器質性月経困難症又は機能性月経困難症と診断された日本人患者
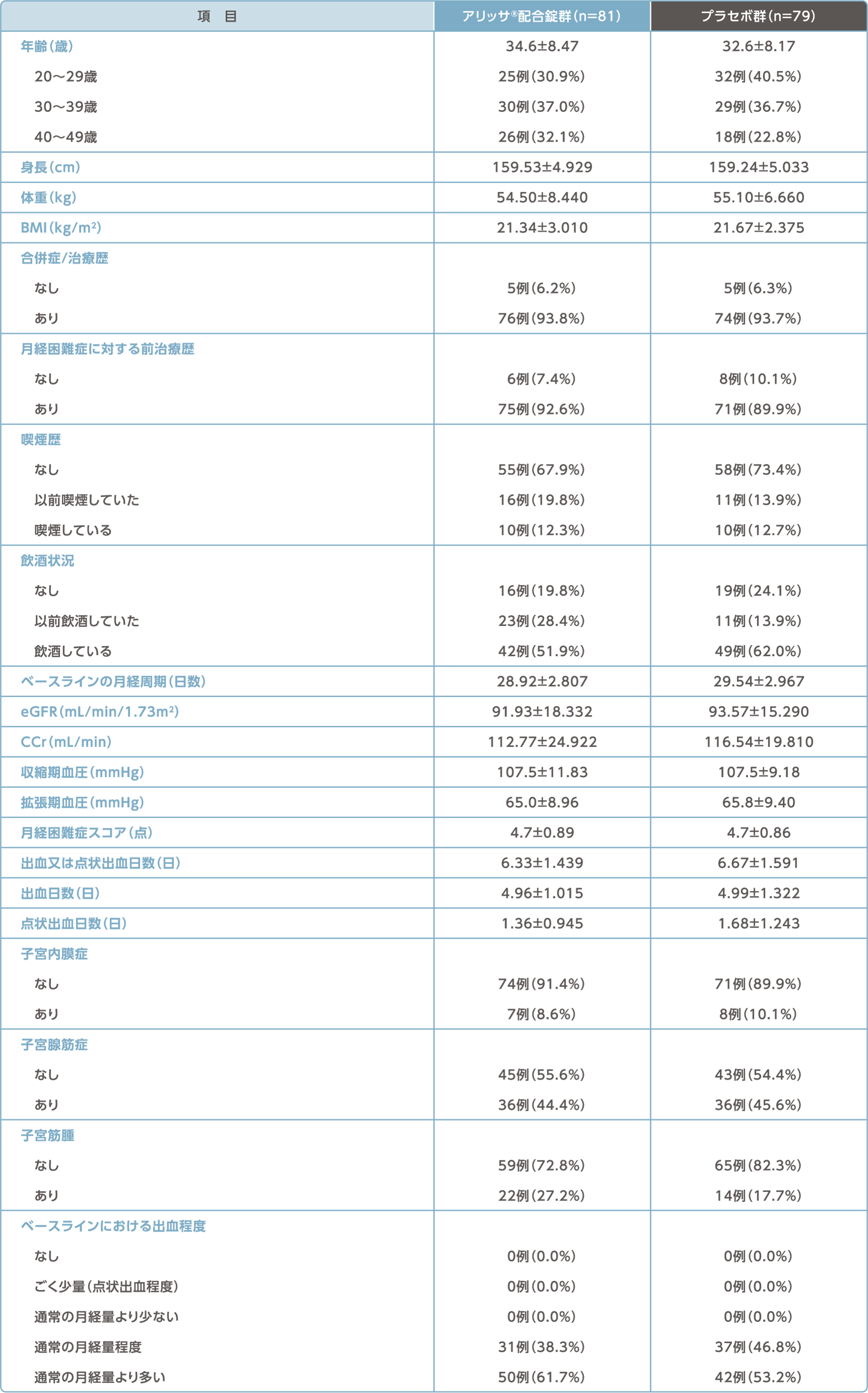
被験者の内訳/被験者背景
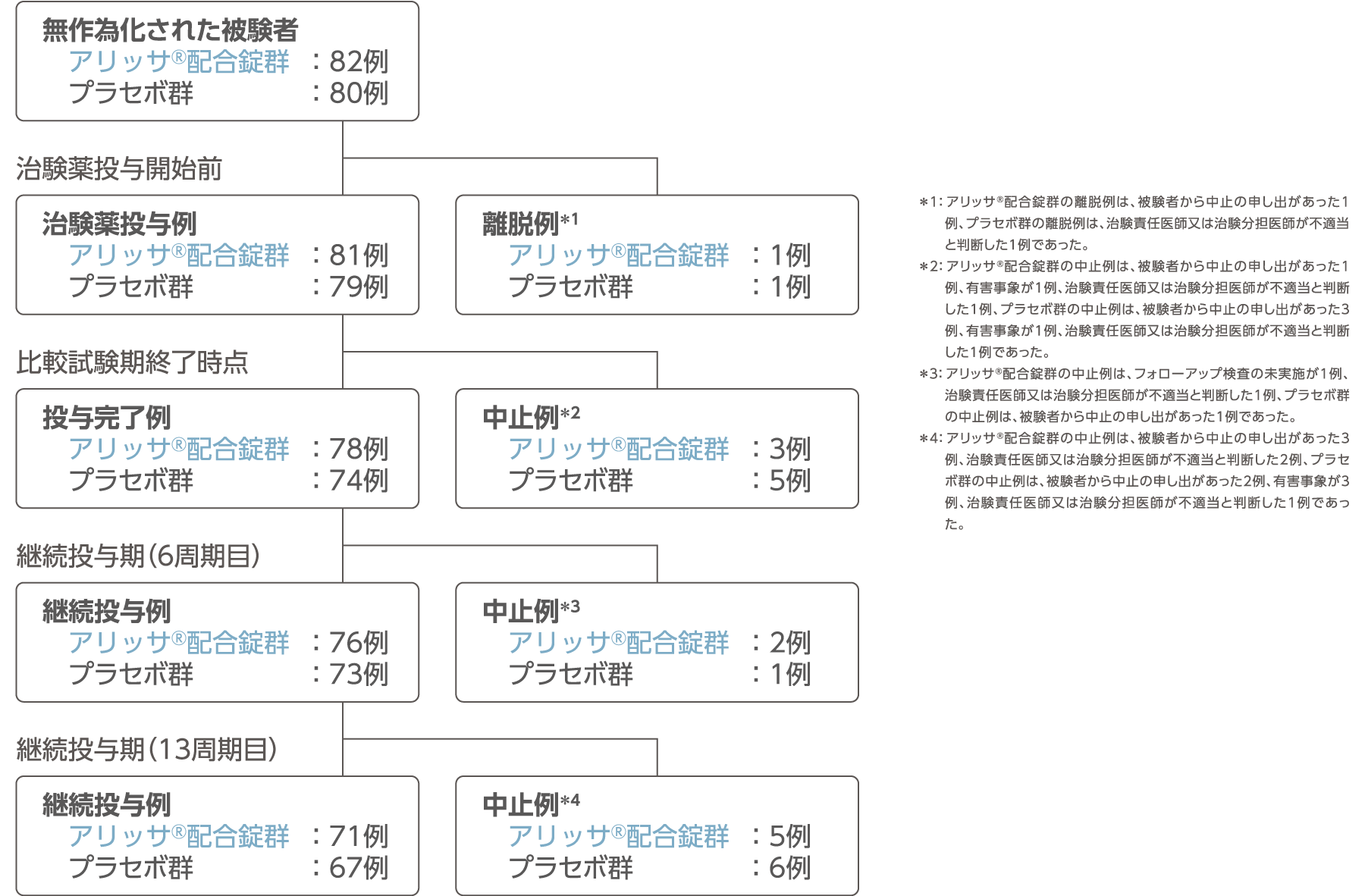
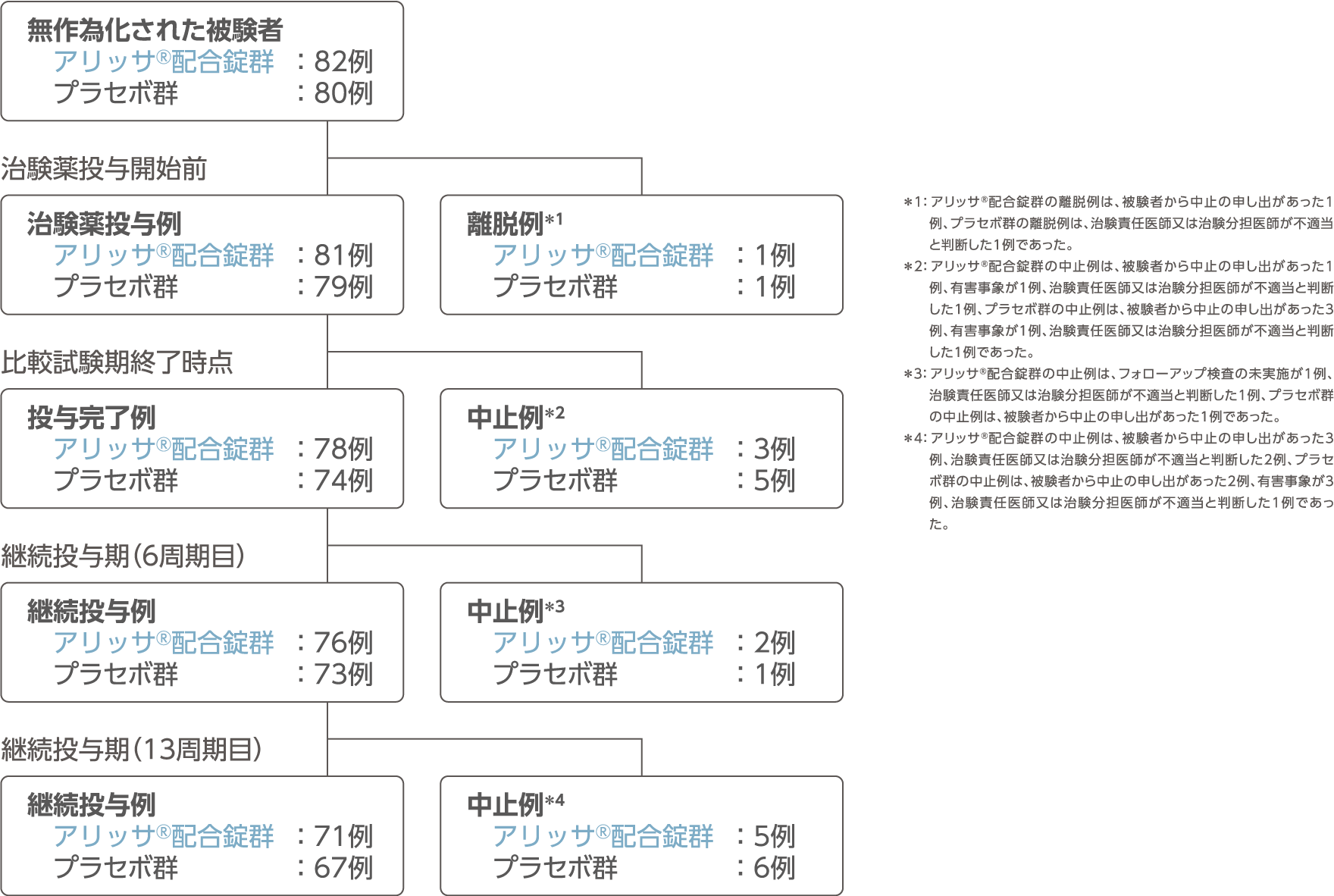
被験者の内訳
被験者背景(FAS)
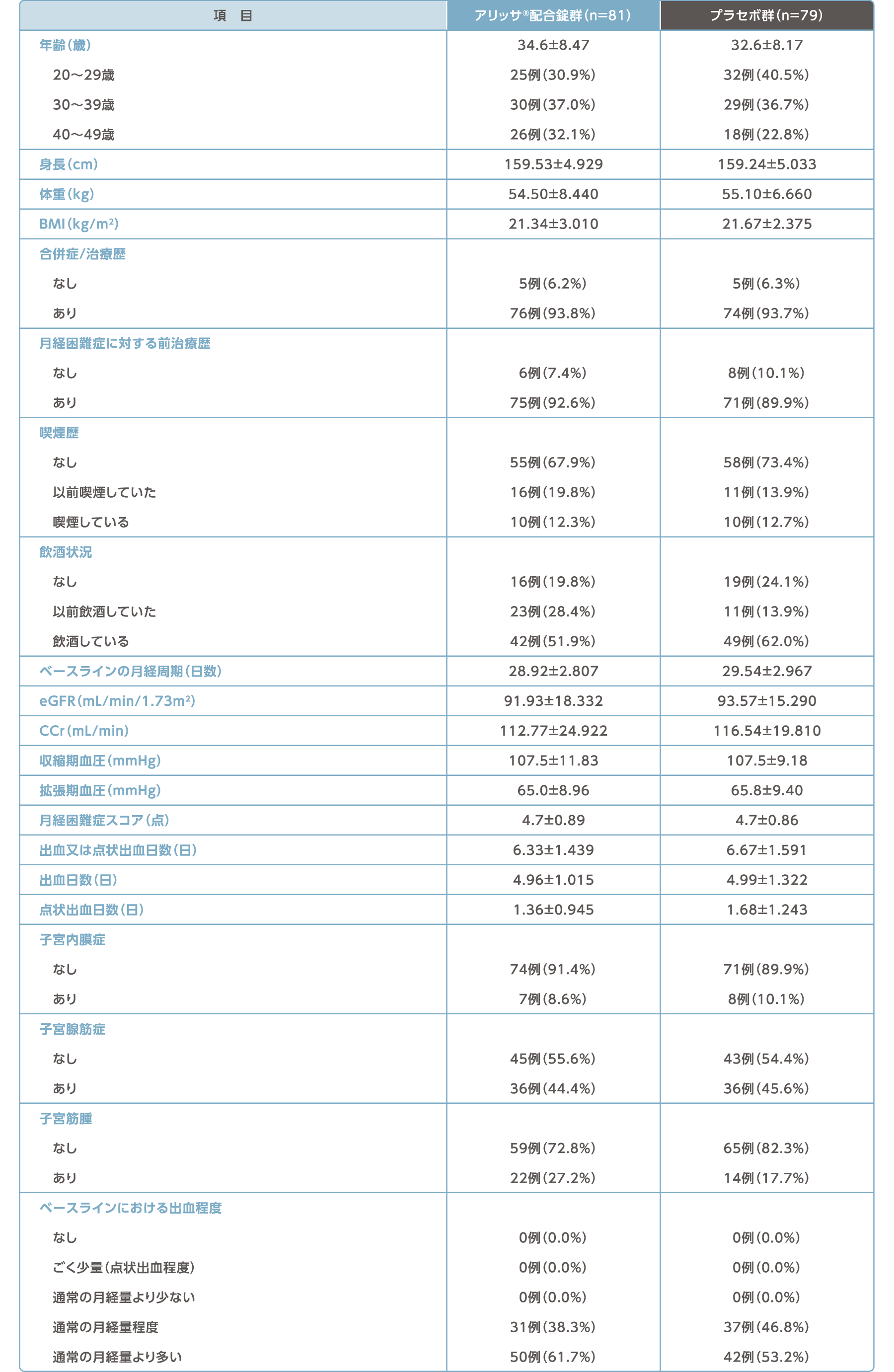
平均値±標準偏差
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
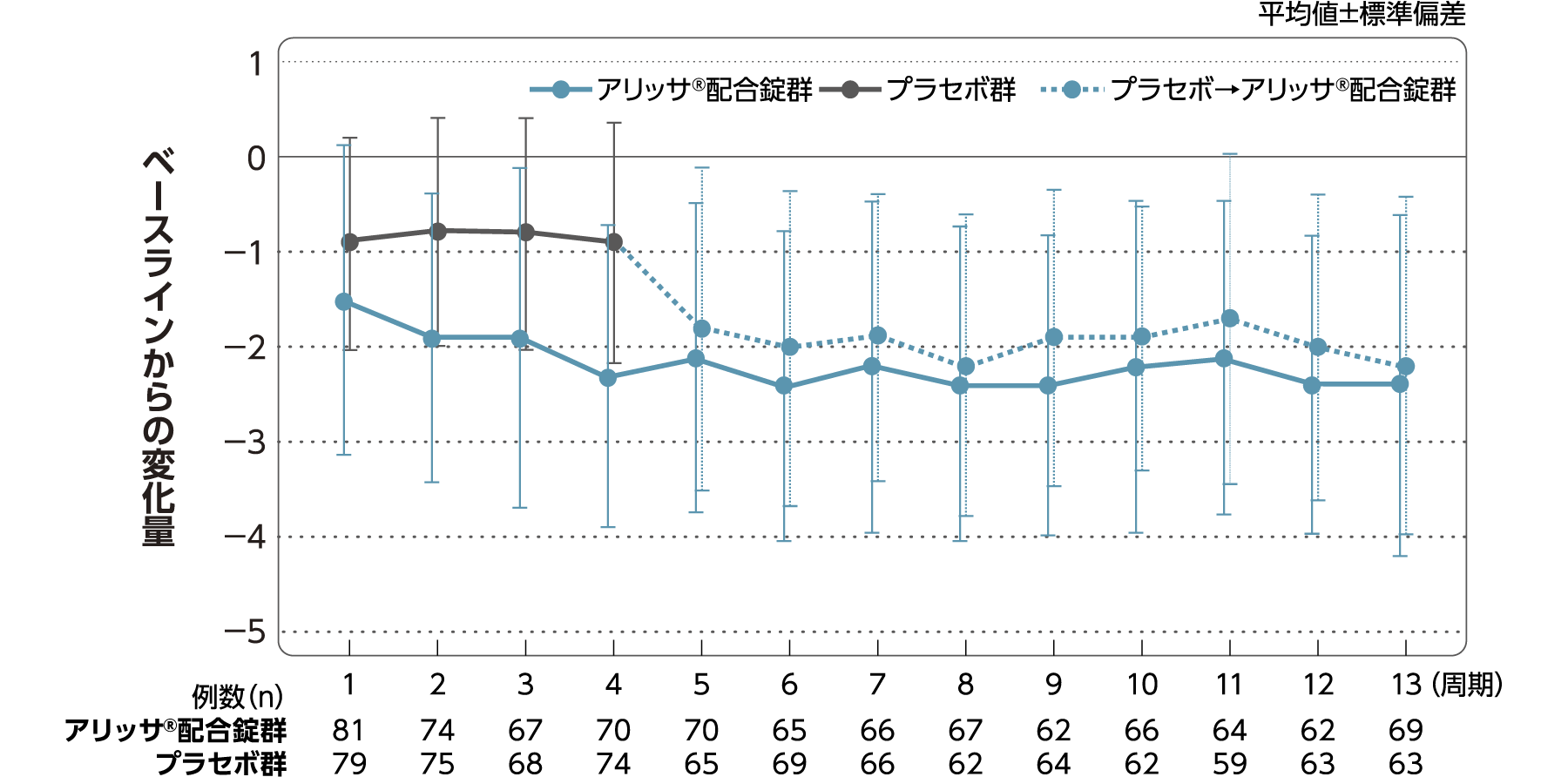
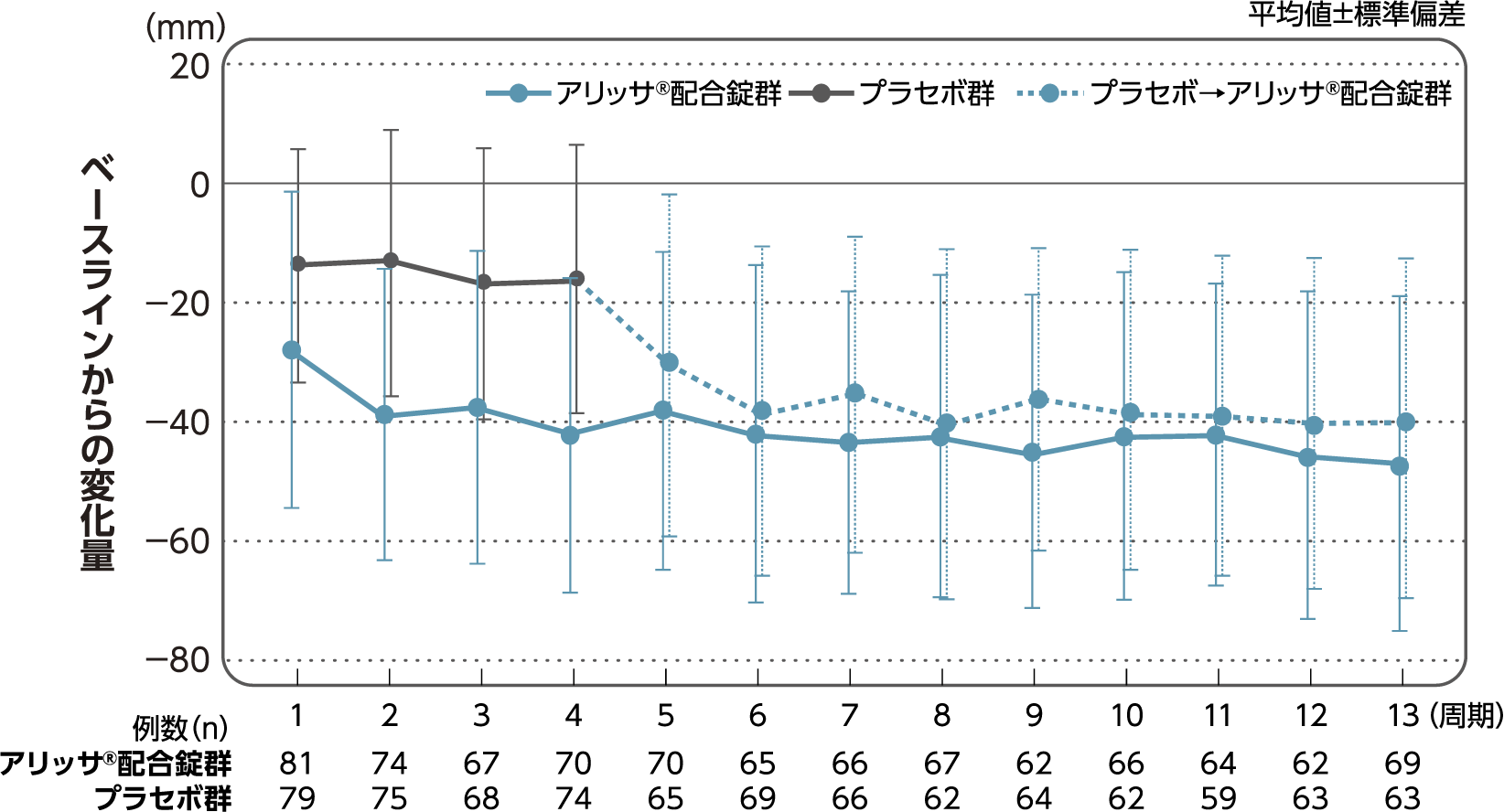
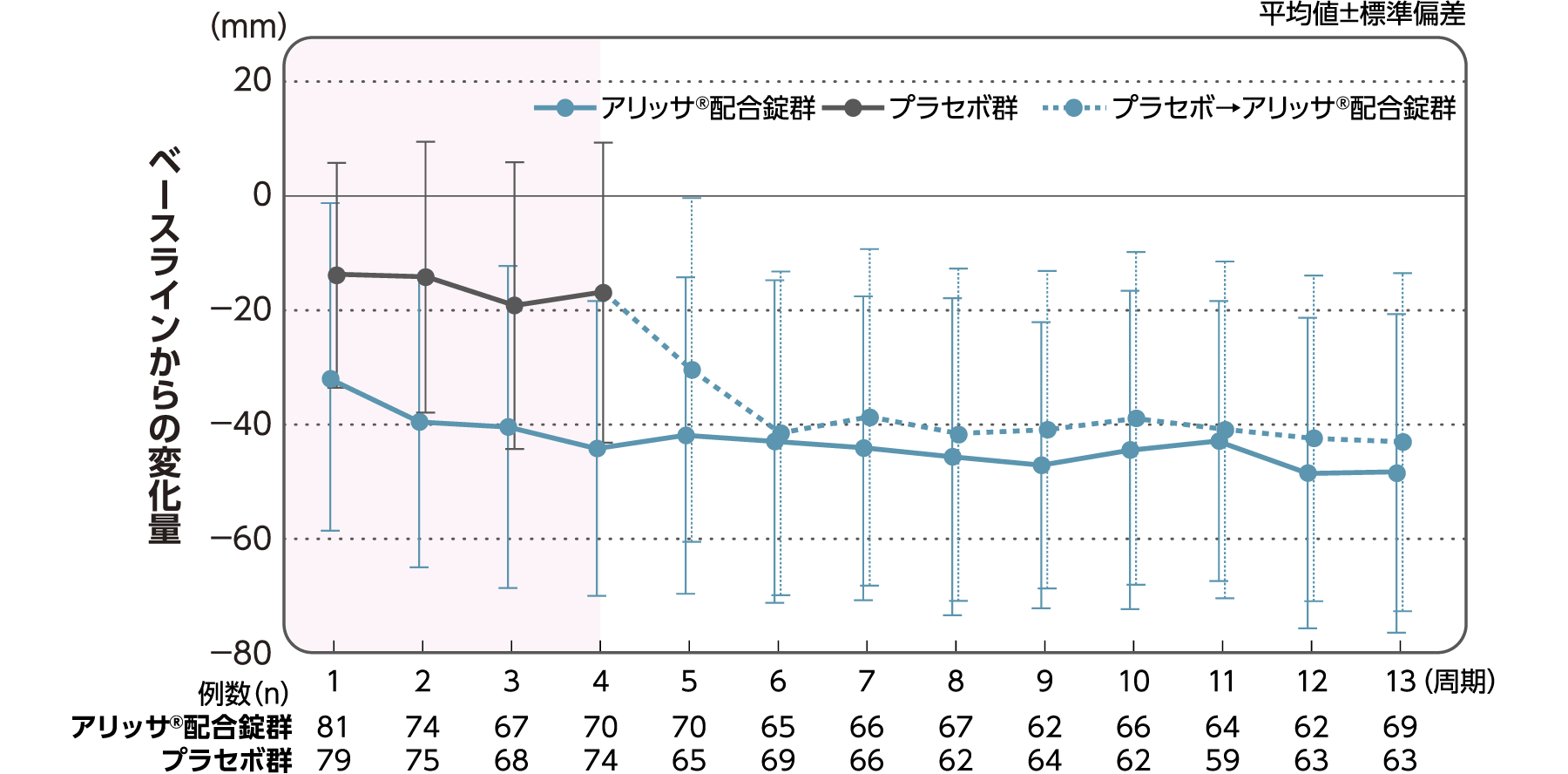
月経困難症スコア
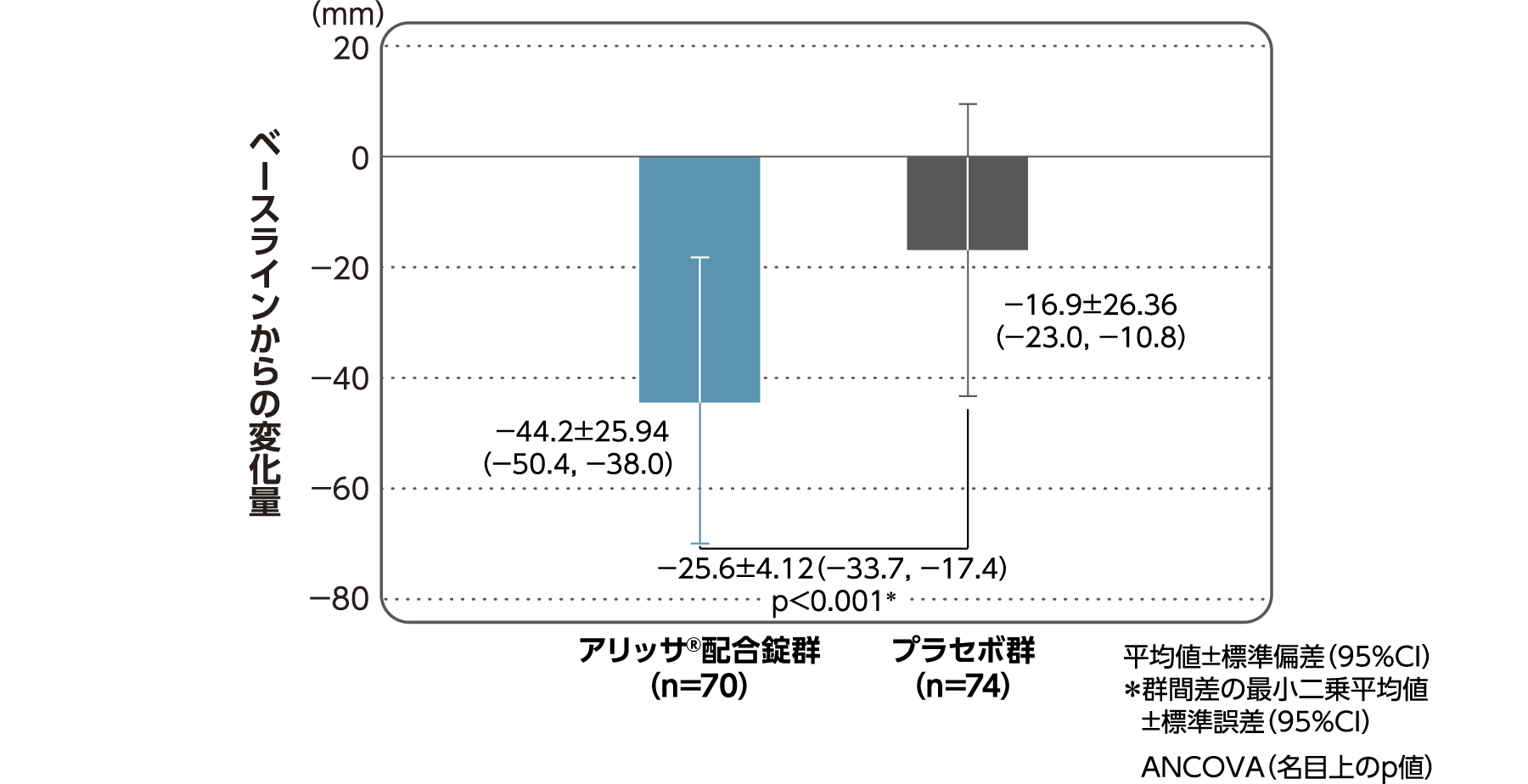
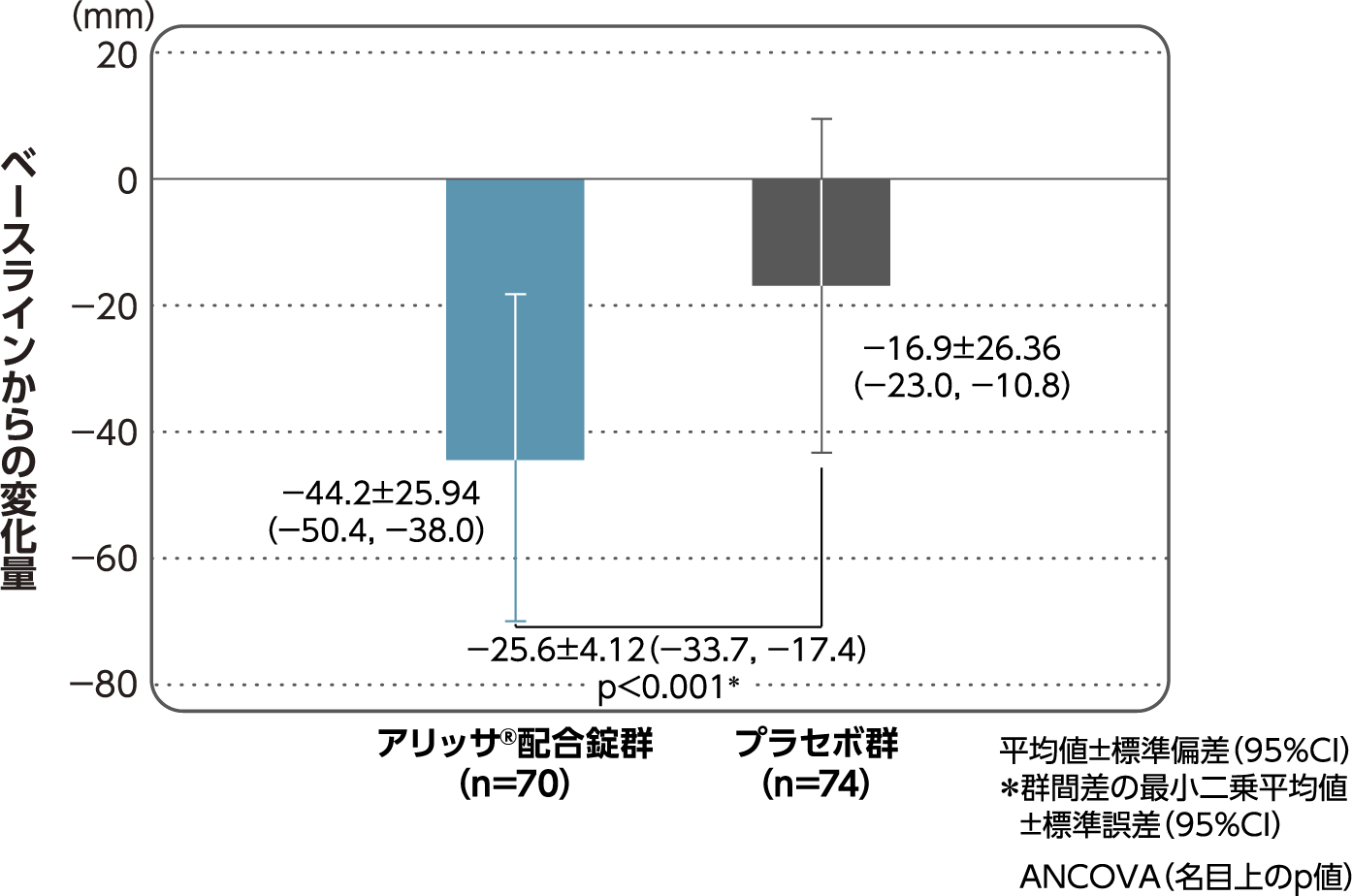
月経困難症スコアのベースラインからの変化量(FAS):
主要評価項目(検証的な解析結果:4周期目)、副次評価項目
月経困難症スコアのベースラインから4周期目までの変化量はアリッサ®配合錠群のプラセボ群に対する優越性が検証された(p<0.001、ANCOVA)[主要評価項目]。
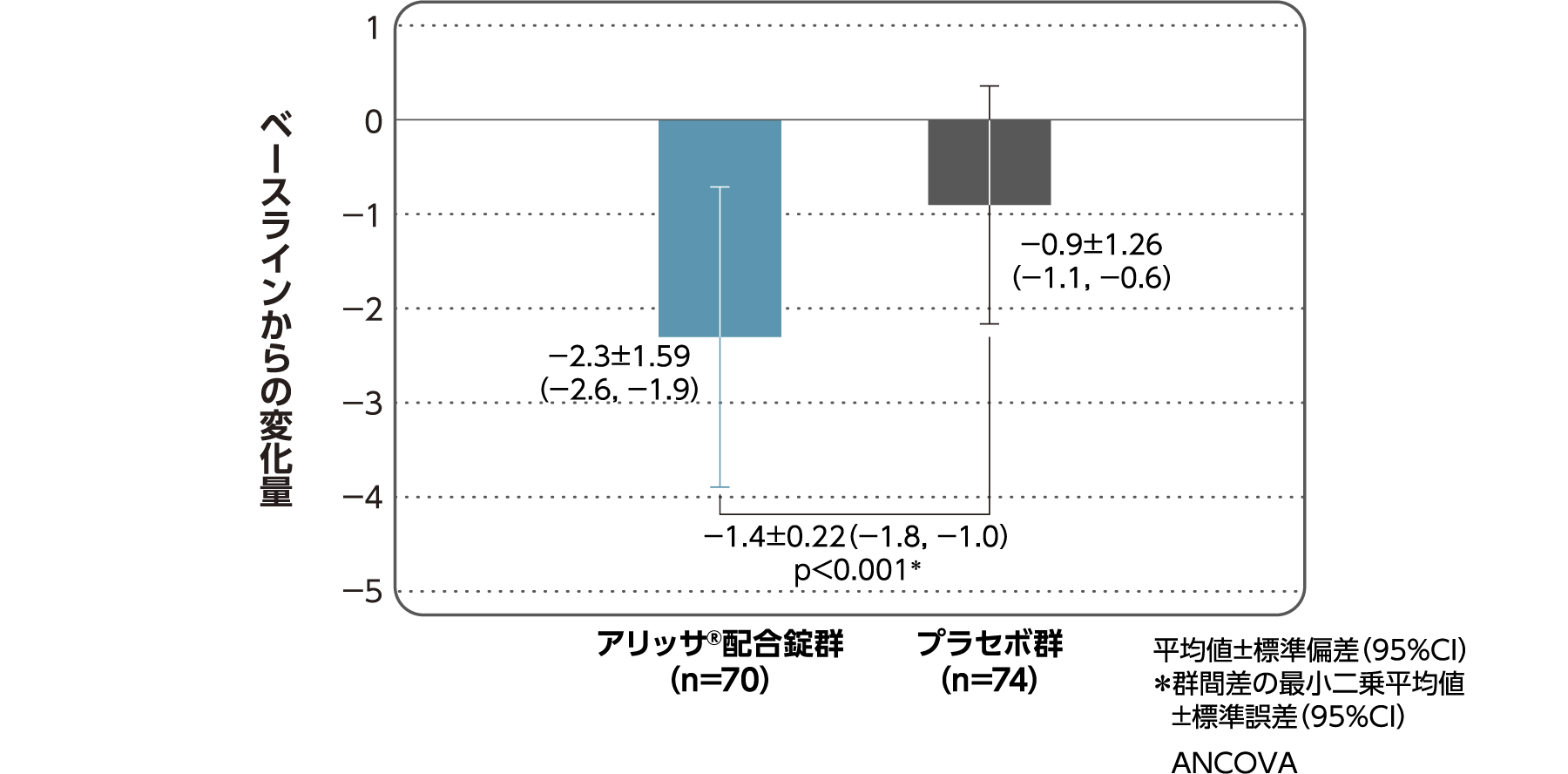
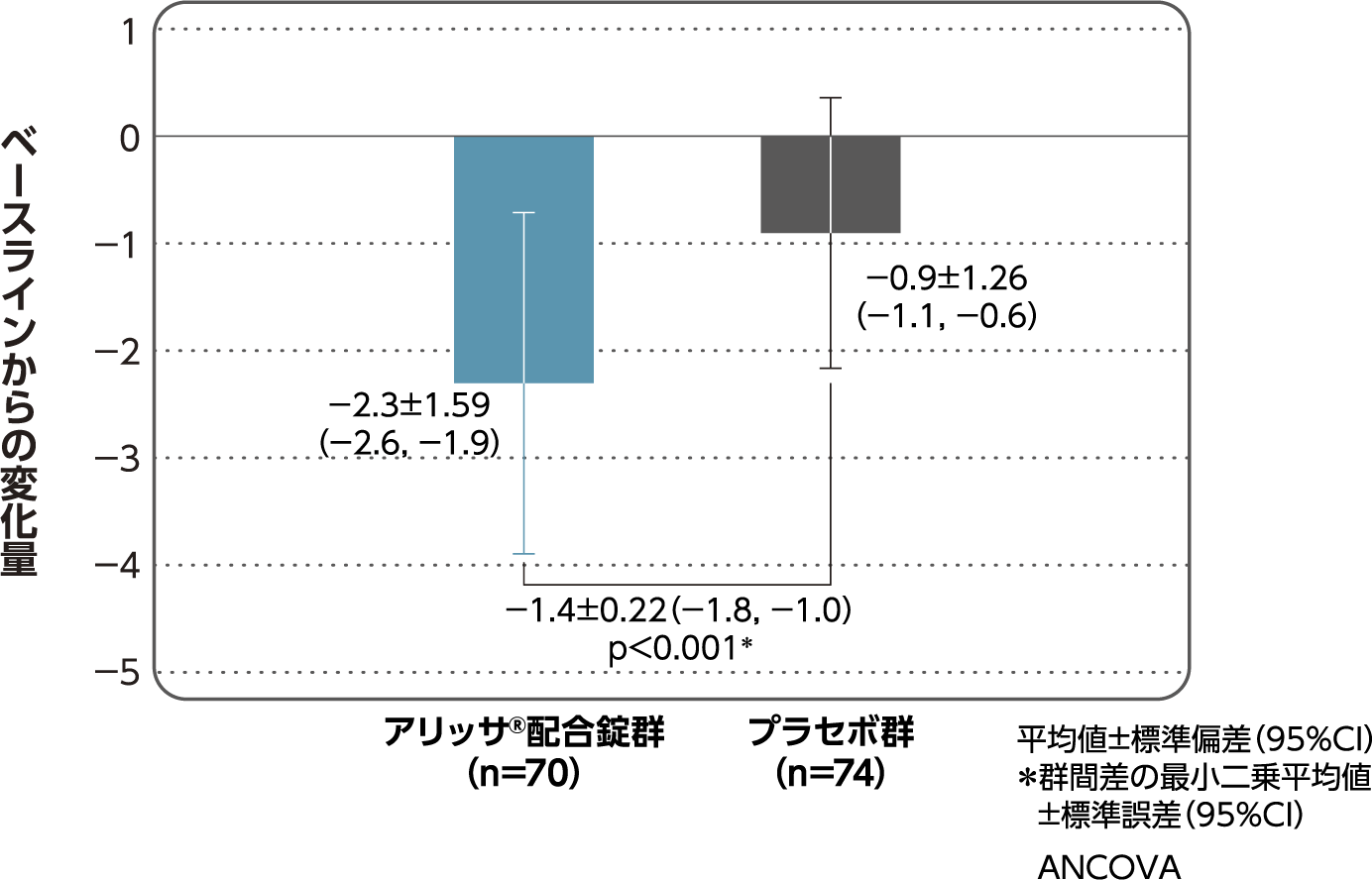
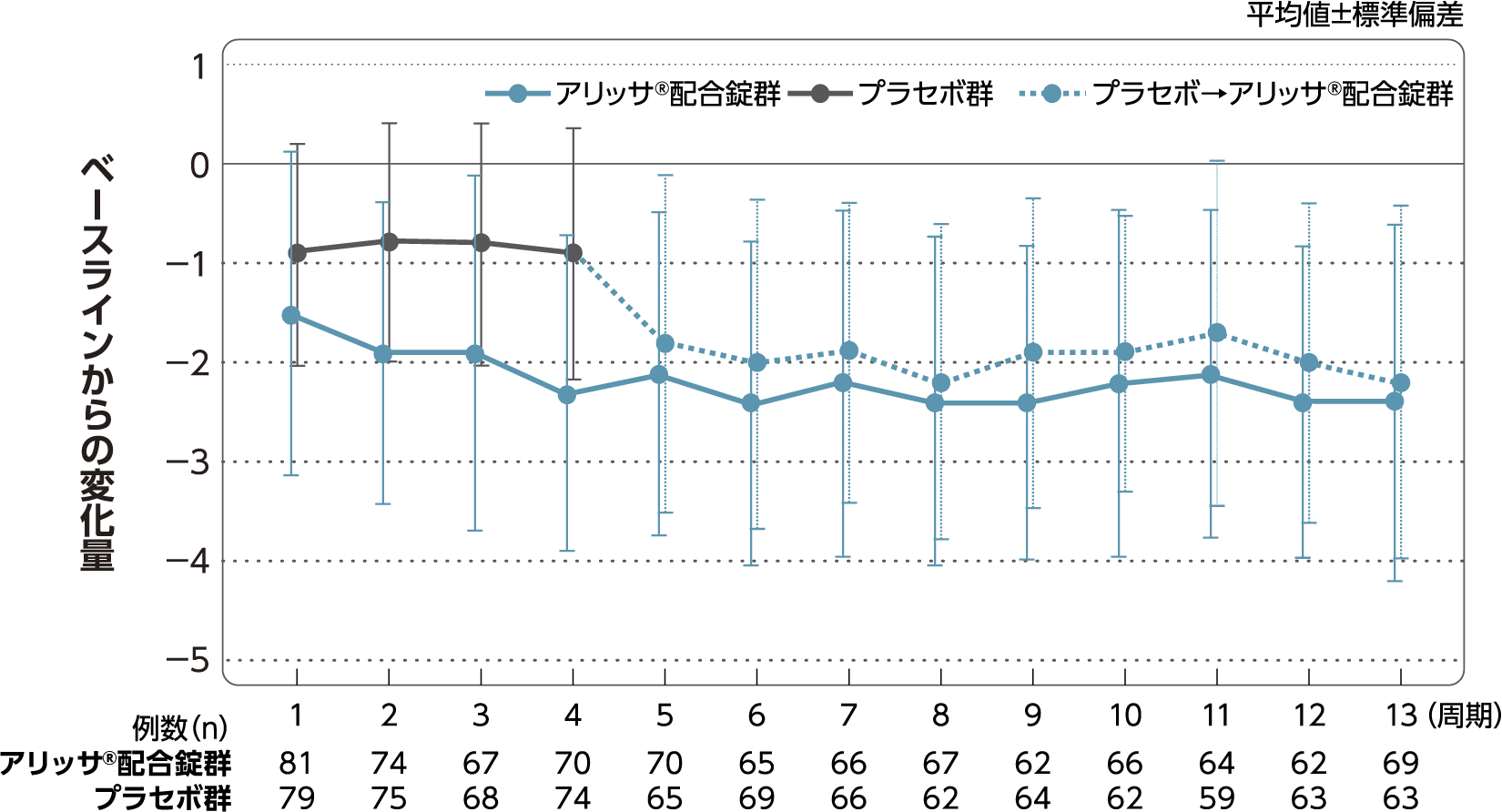
- ANCOVA:
- 4周期目及び4周期目までに投与中止に至った時点での月経困難症スコアの変化量(投与後-ベースライン)を従属変数、投与群及び月経困難症診断カテゴリーを固定効果、ベースラインの月経困難症スコアを共変量とした。
欠測値はLOCFによって補完した。有意水準:2.5%(片側)、群間差:アリッサ®配合錠群-プラセボ群
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
月経困難症症状に対するVAS値
月経困難症症状に対するVAS値のベースラインからの変化量(FAS):副次評価項目
月経困難症症状に対するVAS値のベースラインから4周期目までの変化量はアリッサ®配合錠群のプラセボ群に対する統計学的な有意差が認められた(p<0.001、名目上のp値、ANCOVA)。
- ANCOVA:
- 4周期目及び4周期目までに投与中止に至った時点での月経困難症症状に対するVAS値の変化量(投与後-ベースライン)を従属変数、投与群及び月経困難症診断カテゴリーを固定効果、ベースラインの月経困難症症状に対するVAS値を共変量とした。
欠測値はLOCFによって補完した。有意水準:2.5%(片側)、群間差:アリッサ®配合錠群-プラセボ群
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
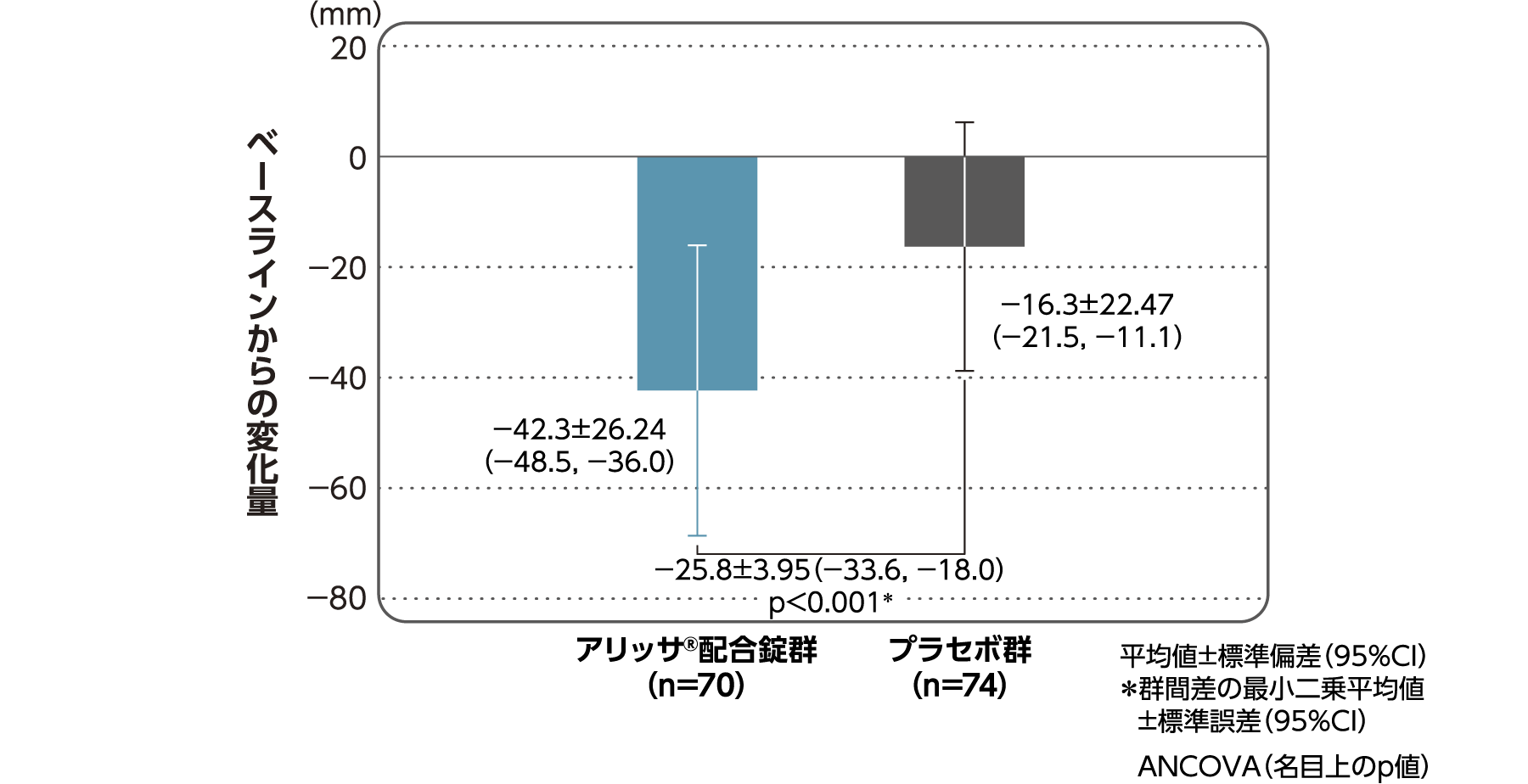
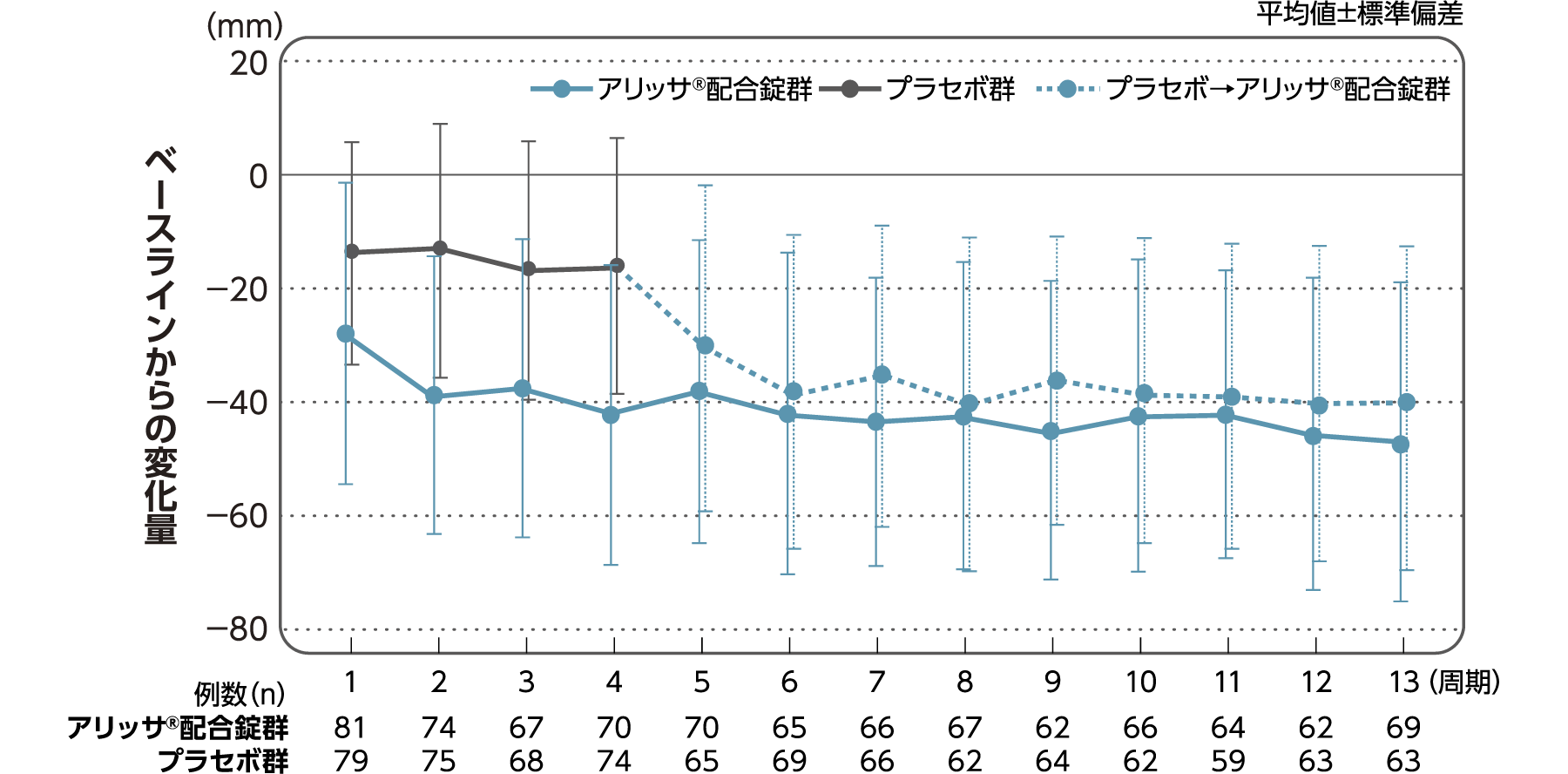
月経時又は消退出血時の骨盤痛(下腹部痛・腰痛)VAS値
月経時又は消退出血時の骨盤痛(下腹部痛・腰痛)VAS値のベースラインからの変化量(FAS):副次評価項目
月経時又は消退出血時の骨盤痛(下腹部痛・腰痛)VAS値のベースラインから4周期目までの変化量はアリッサ®配合錠群のプラセボ群に対する統計学的な有意差が認められた(p<0.001、名目上のp値、ANCOVA)。
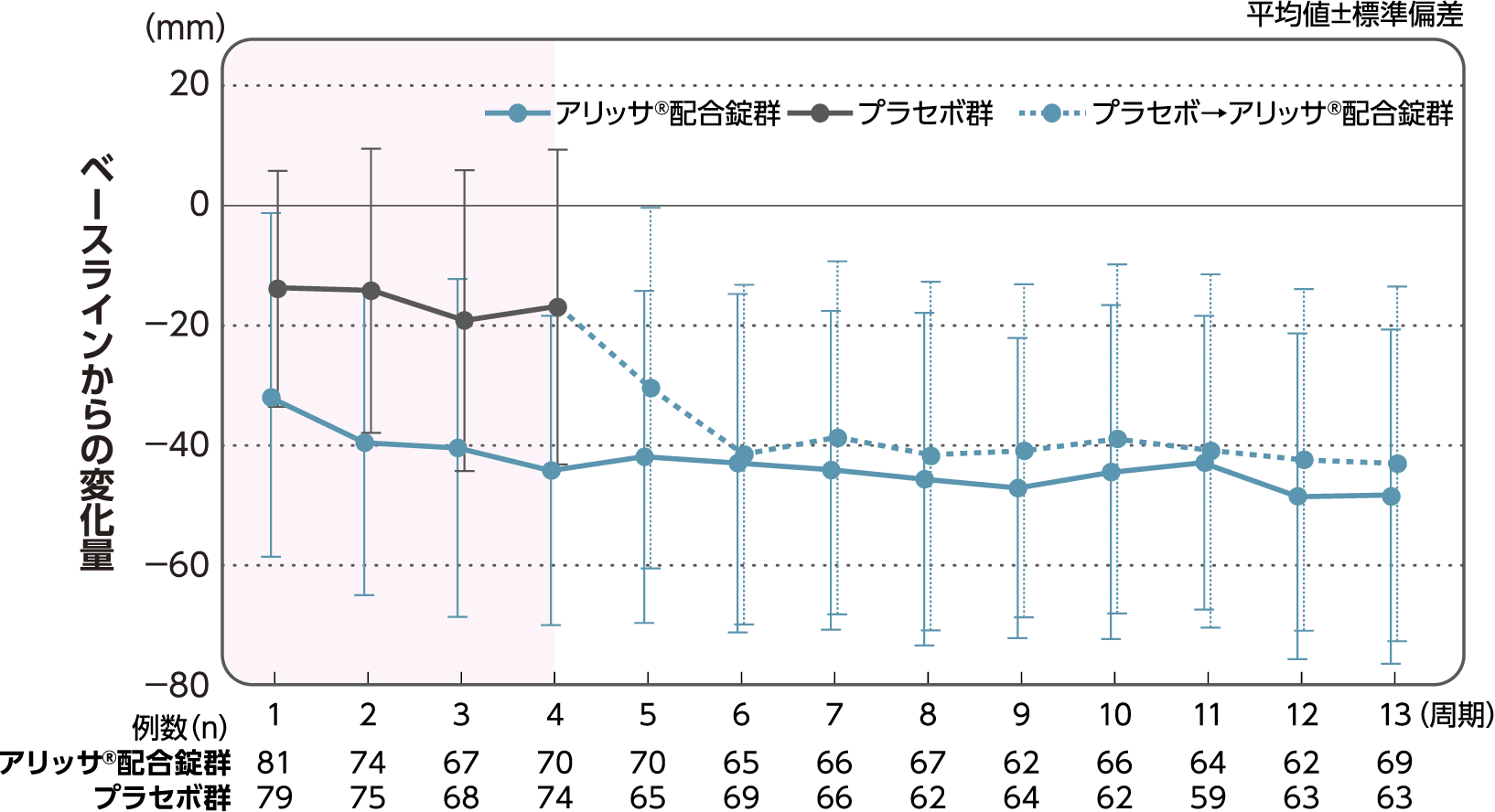
- ANCOVA:
- 4周期目及び4周期目までに投与中止に至った時点での骨盤痛VAS値の変化量(投与後-ベースライン)を従属変数、投与群及び月経困難症診断カテゴリーを固定効果、ベースラインの骨盤痛VAS値を共変量とした。
欠測値はLOCFによって補完した。有意水準:2.5%(片側)、群間差:アリッサ®配合錠群-プラセボ群
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
安全性
副作用(SAS):安全性評価項目
4周期目終了時点までにアリッサ®配合錠群で64/81例(79.0%)、プラセボ群で28/79例(35.4%)に、 また13周期目終了時点までに、アリッサ®配合錠群で76/81例(93.8%)、プラセボ→アリッサ®配合錠群*で67/74例(90.5%)に副作用が認められた。
(1)主な副作用(いずれかの群で発現割合が5%以上)
4周期目終了時点までに、アリッサ®配合錠群では月経中間期出血49/81例(60.5%)、重度月経出血12/81例(14.8%)、悪心8/81例(9.9%)、乳房痛6/81例(7.4%)、頭痛3/81例(3.7%)が、プラセボ群では、月経中間期出血13/79例(16.5%)、頭痛7/79例(8.9%)、乳房痛1/79例(1.3%)が認められた。
また、13周期目終了時点までに、アリッサ®配合錠群では月経中間期出血64/81例(79.0%)、重度月経出血19/81例(23.5%)、悪心8/81例(9.9%)、乳房痛6/81例(7.4%)、頭痛及び骨盤痛各5/81例(6.2%)、倦怠感及びフィブリンDダイマー増加各2/81例(2.5%)、希発月経1/81例(1.2%)が、プラセボ→アリッサ®配合錠群*では、月経中間期出血52/74例(70.3%)、頭痛9/74例(12.2%)、悪心及び希発月経各8/74例(10.8%)、重度月経出血及び乳房痛各7/74例(9.5%)、倦怠感及びフィブリンDダイマー増加各4/74例(5.4%)、骨盤痛3/74例(4.1%)が認められた。
(2)重篤な有害事象
4周期目終了時点までの治験薬投与期間中に重篤な有害事象は認められなかった。
13周期目終了時点までに、アリッサ®配合錠群で、統合失調感情障害及び物質使用障害が各1例1件、プラセボ→アリッサ®配合錠群*で、トリプルネガティブ乳癌が1例1件に重篤な有害事象が認められた。
(3)投与中止に至った有害事象
4周期目終了時点までに、アリッサ®配合錠群で片頭痛が1例1件、プラセボ群で片頭痛及び倦怠感が各1例1件に投与中止に至った有害事象が認められた。
13周期目終了時点までに、アリッサ®配合錠群で片頭痛が1例1件、プラセボ→アリッサ®配合錠群*で、貧血、フィブリンDダイマー増加、可溶性フィブリンモノマー複合体増加、前兆を伴う片頭痛及び月経中間期出血が各1例1件に投与中止に至った有害事象が認められた。
(4)死亡に至った有害事象
本治験では死亡例は両群共に認められなかった。
(5)特に注目すべき有害事象
4周期目終了までに、血栓症に関連する有害事象、DRSP投与に関連すると考えられる有害事象及び器質性疾患に関連する有害事象は認められなかった。EP配合剤投与に関連すると考えられるその他の有害事象として、片頭痛がアリッサ®配合錠群で1例1件、プラセボ群で2例2件に認められた。
13周期目終了時点までに、血栓症に関連する有害事象、DRSP投与に関連すると考えられる有害事象及び器質性疾患に関連する有害事象は認められなかった。EP配合剤投与に関連すると考えられるその他の有害事象として、アリッサ®配合錠群で、片頭痛が1例1件、プラセボ→アリッサ®配合錠群*で、前兆を伴う片頭痛が1例1件に認められた。
*5周期目にプラセボからアリッサ®配合錠投与に切り替え。切り替え後の36週間のデータのみを集計。
MedDRA ver. 24.0
(6)各投与周期における有害事象として報告された月経中間期出血の程度別の発現割合
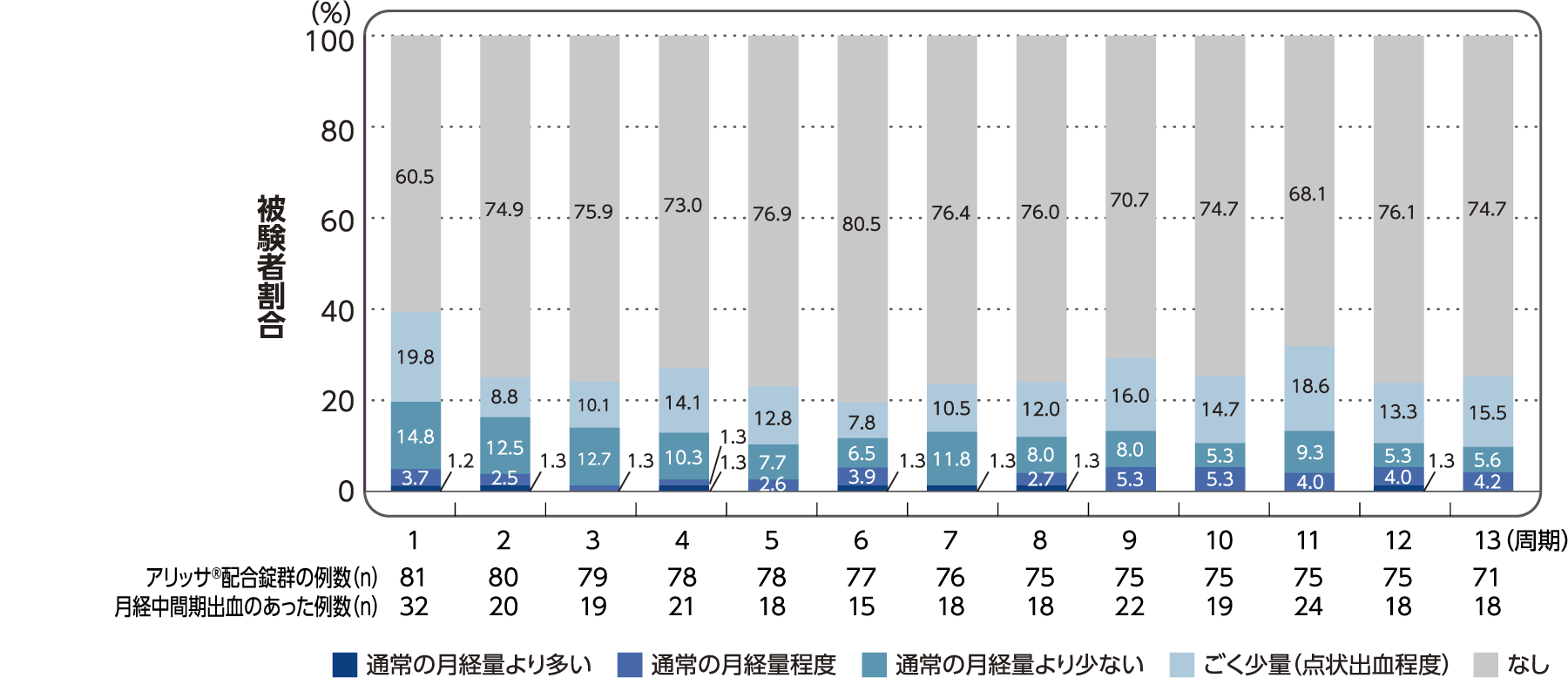
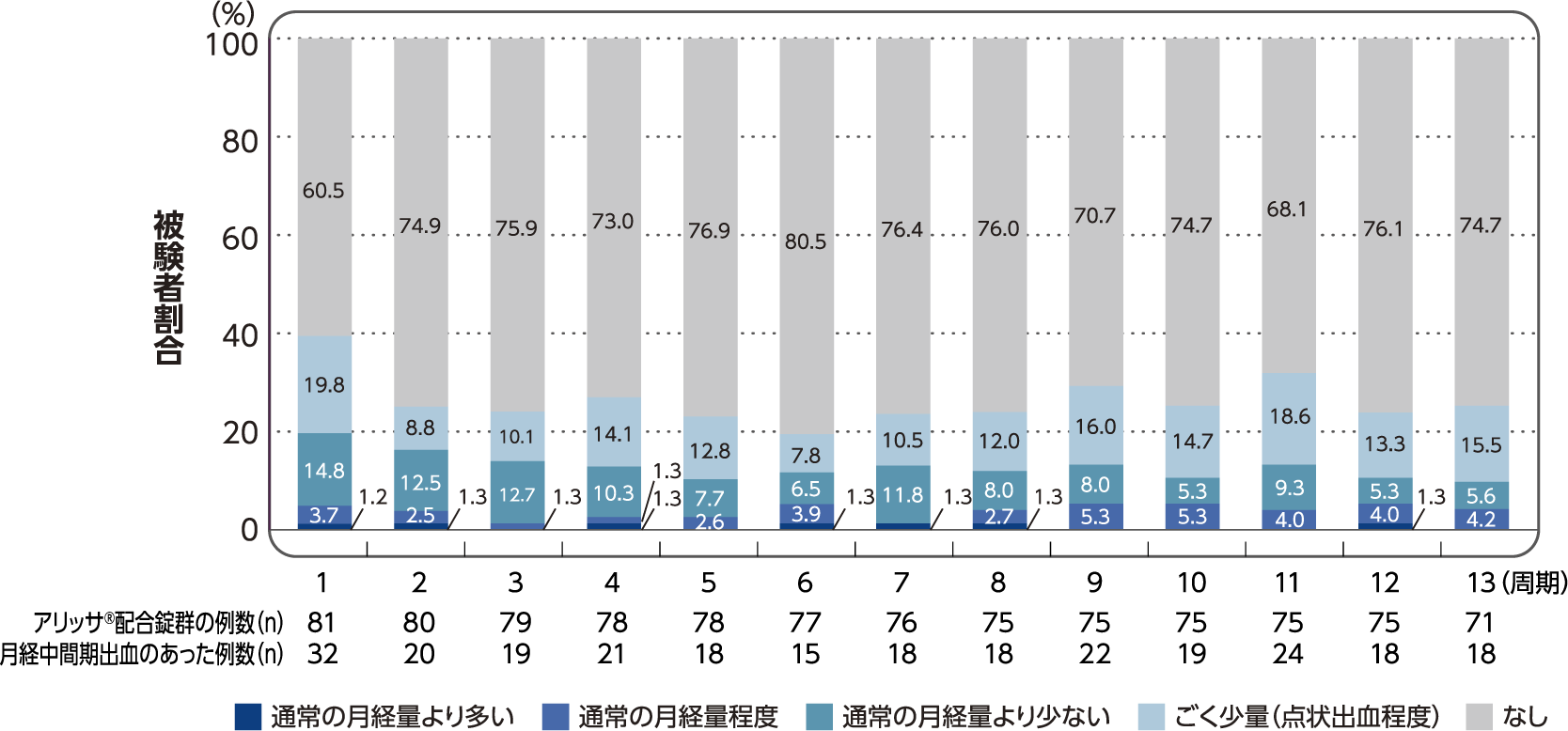
社内資料:月経困難症患者を対象とした国内第III相試験/FSN-013P-03(承認時評価資料)
国内臨床試験で使用された評価の定義とスケジュール
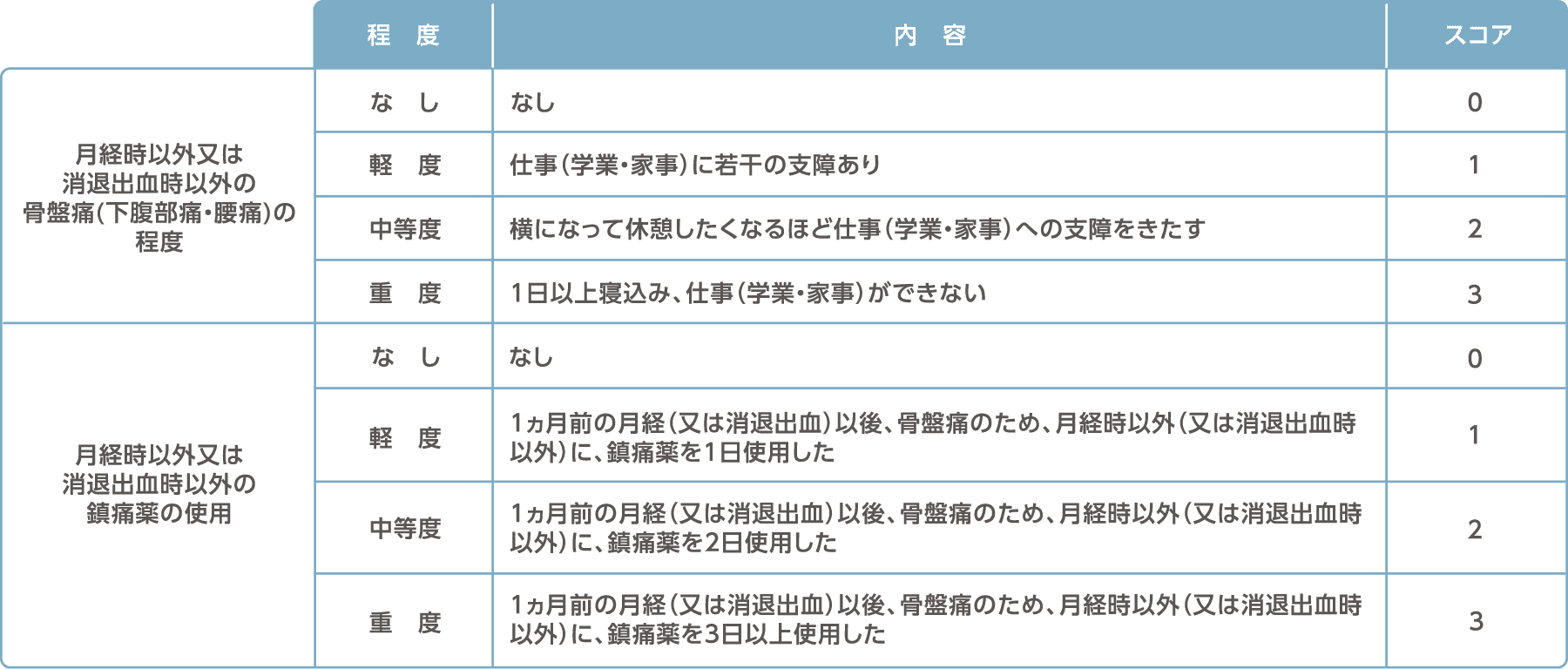
月経困難症スコア

月経時以外又は消退出血時以外の骨盤痛スコア
VAS
Visual Analogue Scale:視覚的アナログスケール
「症状又は痛みがない:0」~「想像できる最大の症状又は痛み:100mm」としたときに、その日に認められた症状の強さ又は痛みの程度を被験者自身の評価により示す
